Docking Preparation Shenanigans in Autodock/Autodock-GPU/Vina
Table of Contents
- Packages
- Downloading the protein and processing it.
- Remove ligands and stuff and only keep proteins
- Download ADFR Suite
- Processing Ligands from SMILES
Autodock likes its protein and ligand file in PDBQT formats, which is sort of a pain to do if you are on a MacOS machine. Thus everything from here is Linux related. I have been very lucky to get access to an HPC, but you should be able to do this on a Raspberry Pi or even PythonAnywhere as they have access to bash!
Packages
The pip commands to install some of the packages.
Downloading the protein and processing it.
Download the protein. 5N2S can be replaced by your PDB code
In our case the receptor is attached to a cytochrome and I would very much like it gone. I don't really need any of the cytochrome fluff because that is way outside the ligand binding pocket. We hit a problem again. Usually the cytochrome kind of things are in a different chain. Here there are not. You can use the 3D viewer of the RCSB page and figure which residues are worth removing, and then come back to this code.
=
=
=
= # Residue IDs worth removing
If you double click on any of the residues, the ID comes up on the right.
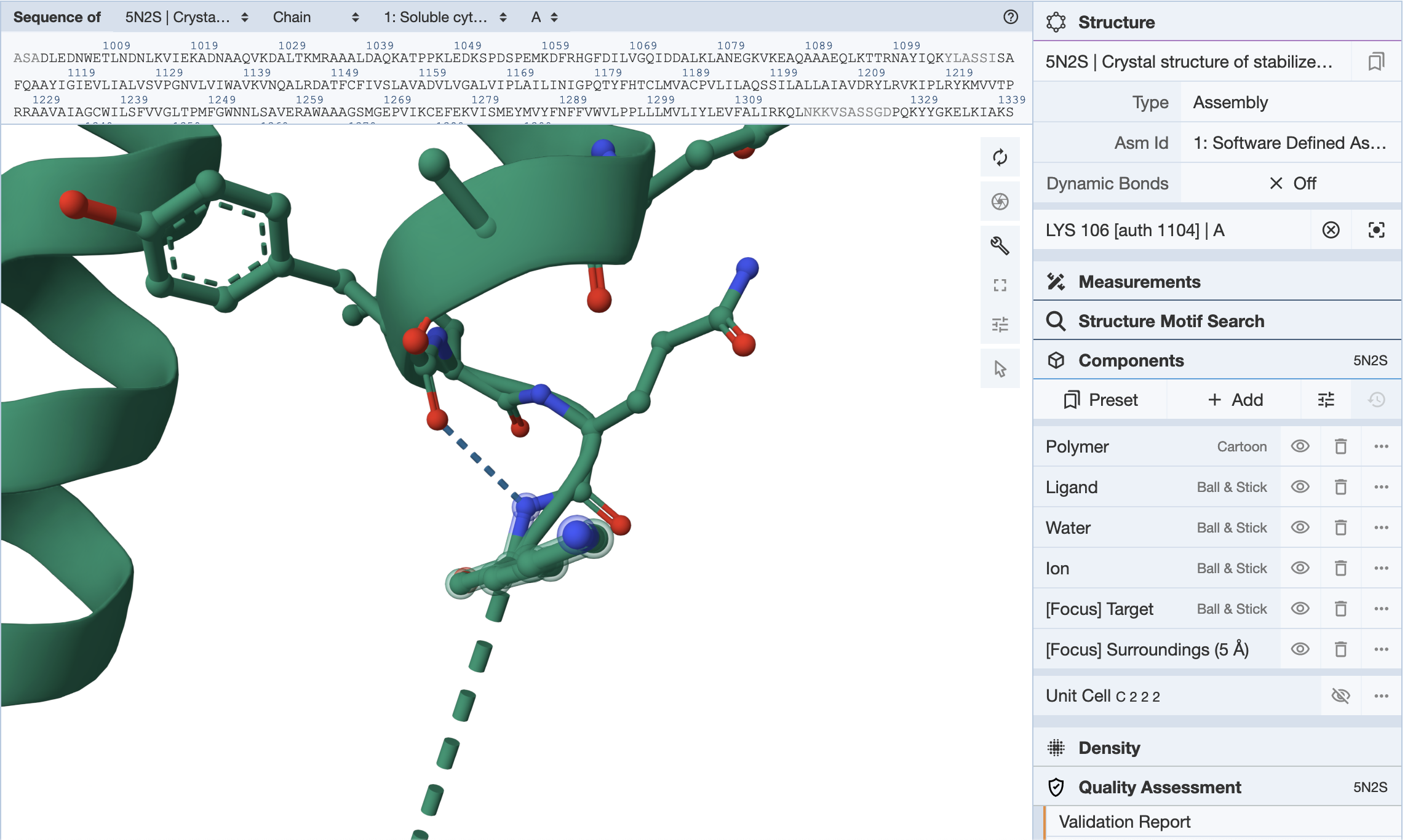
Remove ligands and stuff and only keep proteins
grep is a good text editing magic thing in unix-based systems.
Download ADFR Suite
We need the ADFR suite to automate a few things that I don't want to break.
Sadly enough, Autodock's installation thingy does not work for me on CentOS and it's just easier to download the tarball file and install from there. So that's what we are doing!
After than unzip the file, cd into the uncompressed directory and just run the installation script. At the end of the installation script, there is an alias that you can add to your terminal which makes life easier. All those commands in order :).
Everyone's alias will look different. DON'T Just copy mine. It won't break or anything just you know!
I am adding hydrogens because my structure probably does not come with hydrogens. I am learning as well so if you think mine does not drop me and email.
Processing Ligands from SMILES
I use a nifty little Python package called Meeko and here's the full code for that. The function takes in a SMILES string and spits out a PDBQT file in your desired place.
# Turns the SMILES string into rdkit molecules and protonates it
=
=
=
# Tries to take the rdkit molecule and embed it into 3D space but it's not always for highly flexible compounds.
# You could try to increase the number of tries but at some point do you want to wait 10 hours for one molecule?
# The 3D ligand now is turned into PDBQT string by meeko and then churned into a file
=
=
Now all you need is to run a docking program. There are many tutorials:
I may add one of my own to the pile someday!
Have fun 😊